
Stereoselektive Silylierung chiraler 1-Aminoallylmetall-Verbindungen mit anschließender Oxidativer Desilylierung
Zusammenfassung
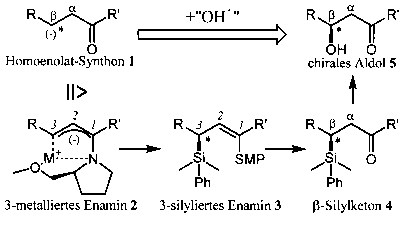
Zahlreiche Naturstoffe und pharmazeutische Wirkstoffe besitzen in b-Position zu einer Carbonylgruppe ein chirales Zentrum. Insbesondere chirale Aldolverbindungen 5 sind weit verbreitet. Neben den klassischen Darstellungsmethoden für chirale Aldolverbindungen 5 stellt die stereoselektive Einführung einer Hydroxylgruppe in ein bestehendes Kohlenstoffgerüst eine interessante Alternative dar. Die formale enantioselektive Knüpfung einer C-O-Bin-dung, ausgehend von dem Homoenolat-Synthon 1, mit einem "Hydroxylkation" entspricht einer zweifachen Umkehrung der natürlichen Polarität der Reaktanden. Dieses Ziel konnte erreicht werden durch die zwischenzeitliche Einführung einer Silylgruppe und anschließender oxidativer Desilylierung. Die Dimethylphenylsilylgruppe besitzt gleich mehrere entscheidende Vorteile: Sie reagiert mit 2 unter hoher Stereoselektivität und hohen chemischen Ausbeuten, sie ist stabil unter den stark basischen Reaktionsbedingungen, sie erleichtert durch ihren a-Effekt eine zweite Metallierung, sie erhöht aufgrund ihres sterischen Anspruchs die Diastereoselektivität einer anschließenden Alkylierung und schließlich lässt sie sich unter vollständiger Retention der Konfiguration in eine Hydroxylgruppe überführen.
Metallierung von 3-Phenyl-substituierten 1-SMP-Aminoallylverbindungen mit t-BuLi oder t-BuLi/KOT ergeben chirale 3-metallierte Enamine 2. Diese dienen als Syntheseäquivalente des Homoenolat-Synthons 1 und werden von Elektrophilen in 3-Position angegriffen. Die Stereoselektivität wird dabei von SMP, einem Prolinderivat, das als chirales Auxiliar dient, induziert. Die Abfangreaktion von 2 mit DMPSCl liefert 3-silylierte Enamine 3. Die Stereo-selektivität der elektrophilen Substitution kann von 64 %ee (S) bis zu 95 %ee (R) am C3-Atom in Abhängigkeit von Lösungsmittel, Metallion und Temperatur variiert werden. Die 3-silylierten Enamine können zu b-Silylketonen 4 hydrolysiert werden, welche wiederum als Vorläufer der chiralen Aldolverbindungen 5 dienen.
Diese Strategie konnte noch durch eine Eintopf-Synthesesequenz verbessert werden. Dabei entstanden durch Metallierung, Silylierung, Metallierung und Alkylierung der einfachen Ausgangsverbindung Allyl-SMP (R=R'=H) fast enantiomerenreine (R)-b-Silylaldehyde. Diese Aldehyde lassen sich durch Oxidieren und anschließender oxidativer Desilylierung zu chiralen b-Hydroxycarbonsäuren überführen oder aber durch Reduzieren und anschließender oxidativer Desilylierung zu chiralen 1,3-Diolen überführen. Nach diesem Konzept aus Silylierung und Alkylierung von Allyl-SMP und anschließender oxidativer Spaltung der C-Si-Bindung ist es möglich eine ganze Bibliothek aus hoch enantiomerenreinen chiralen Aldolverbindungen darzustellen.
|
|